Sickle Cell Disease
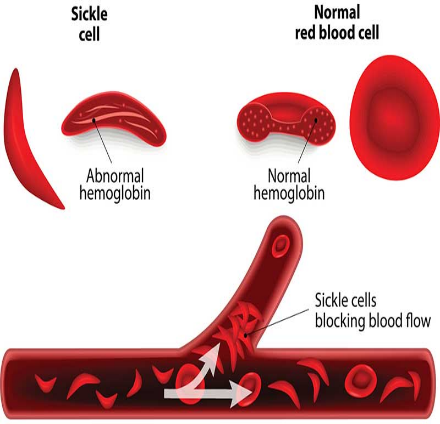
Sickle cell disease (SCD) is a group of inherited (genetic) conditions which affects the red blood cells in the blood.
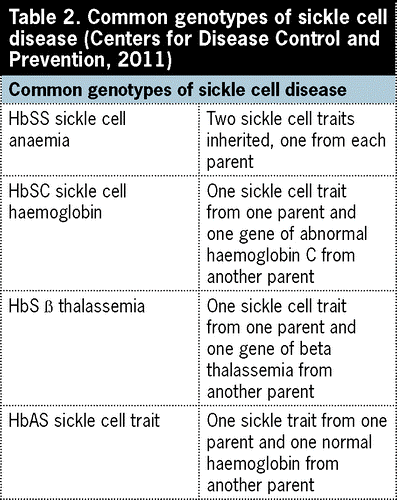
Sickle cell anaemia is the name of a specific form of SCD in which there are two sickle cell genes.
There are several types of sickle cell disease- Sickle Cell Anaemia (SS), Sickle Haemoglobin-C Disease (SC), Sickle Beta-Plus Thalassemia, Sickle Beta-Zero Thalassemia, Haemoglobin SD, Haemoglobin SE, And Haemoglobin SO.

EPIDEMIOLOGY
Sickle cell disease and trait affects millions of people globally and is particularly common among those whose ancestors come from sub-Saharan Africa.
About 5–7% of the global population carries an abnormal haemoglobin gene. The most predominant form of haemoglobinopathy worldwide is sickle cell anemia. These conditions are common in regions that have widespread malaria.
The sickle cell trait (patients with one sickle cell gene) has resistance against malaria, particularly falciparum malaria. Sickle cell was used as a method to decrease the deaths associated with malaria.
The prevalence of sickle cell trait ranges between 10 and 45% in various parts of sub-Saharan Africa. In Nigeria, carrier prevalence is about 20 to 30%. SCA affects about 2 to 3% of the Nigerian population of more than 160 million.
Sickle cell is also more common in:
Hispanics from Central, South America and the Caribbean
People of Middle Eastern (Saudi Arabia, Bahrain and Oman) descent
People of Mediterranean (Turkey, Greece and Italy) descent
People of Southeast Asian (India) descent
LET’S GO BACK
In 1874, Dr Horton, a Sierra Leonian medical Doctor, reportedly gave the first description of clinical symptoms and signs which is now referred to as sickle cell disease
Although sickle cell disease was widely known in Africa, it wasn’t discovered in Western medicine until 1910.
Walter Clement Noel was the first person described with sickle cell disease. Noel was a dental student from Grenada, who studied in Chicago. When experiencing a pain episode, Noel sought out medical care at a neighbourhood hospital. Noel was assigned to Dr Ernest Irons who was a medical resident of cardiologist Dr James Herrick.
A BIT OF GENETICS
In sickle cell anaemia there’s a point mutation, which leads to a substitution of valine for glutamate on the 6th amino acid of the beta globin chain.
The glutamate protein molecule, which is hydrophilic, polar, and negatively charged, is replaced by a less polar, hydrophobic, neutral amino acid, valine.
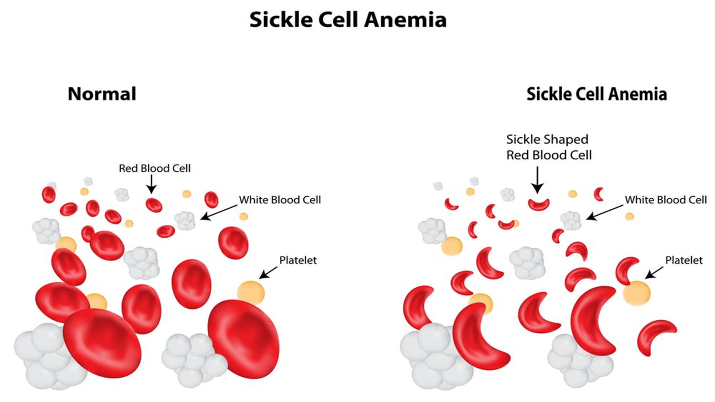
The abnormal biochemistry of this mutant haemoglobin induces polymerization of Hb S molecules within the red cells, which leads to sickling.
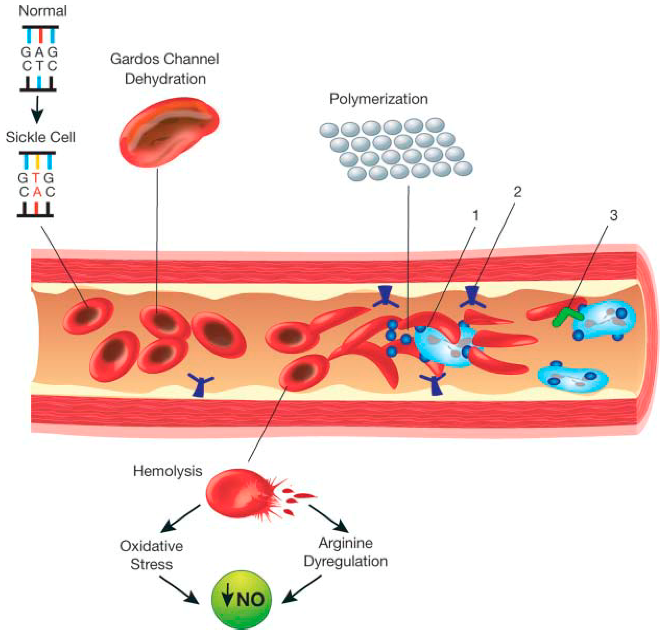
The normal human red blood cell is biconcave enabling it to adjust to the conditions of the body the sickle red blood cell on the other hand, can get trapped within the blood vessels leading to painful crisis experienced by a sickle cell warrior. A sickle cell warrior doesn’t always have a sickled red blood cell, these red blood cells sickle under conditions of stress and dehydration as perceived by the body.
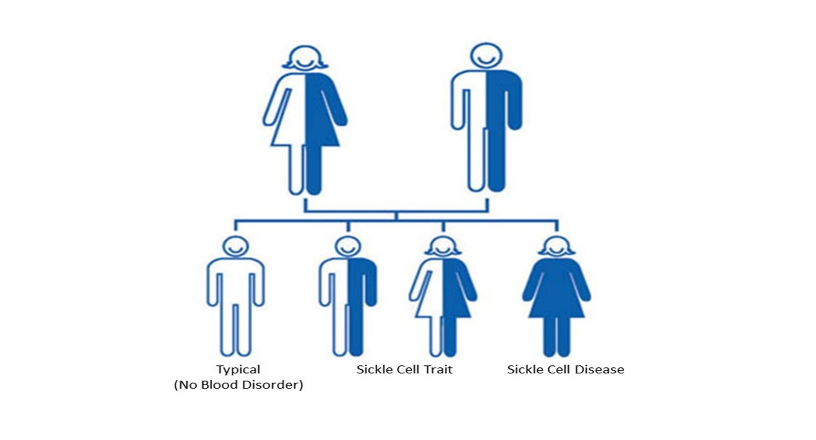
This disease can be inherited when both mating partners are carriers of the Hbs gene, when this happens, they have a ¼ (autosomal recessive inheritance) chance of passing the defective gene to their offspring in each pregnancy.
How does SCA present?
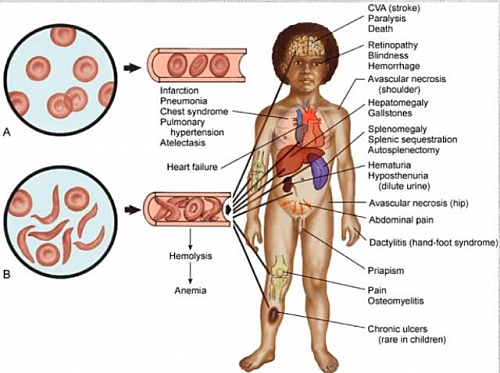
Sickle cell warriors can experience symptoms ranging from painful bones and generalized body aches, acute chest syndrome and even as severe as a stroke.
Signs and symptoms of sickle cell disease are different from person to person and can range from mild to severe. There are many factors that impact how severe sickle cell disease may be—even siblings may not have the same degree of severity.
Sickle cell symptoms start around one year of age; though, with severe sickle cell disease, symptoms can start as early as four to six months of age. One of the first signs a baby has sickle cell disease, other than screening, is dactylitis, which is the swelling of their hands and feet.
In conditions of very high altitude, people with sickle cell trait can present with mild forms of sickle crisis. Chronic kidney disease and a very rare form of kidney cancer (renal medullary carcinoma) are also associated with sickle cell trait.
Diagnostic Testing
During Pregnancy
Testing for sickle cell can be performed during pregnancy. A prenatal test can be conducted to find out if the fetus has sickle cell disease or sickle cell trait by collecting a sample of the amniotic fluid, the fluid around the baby. Chorionic villus sampling, a DNA test, is an additional way to test the baby’s sickle cell genetic status. Additionally, there is also ongoing research to test maternal blood for fetal DNA.
Newborn Screening
Newborn screening consists of a heel-prick blood sample that is then analyzed for abnormal hemoglobin. This test can identify both sickle cell disease and sickle cell trait. Universal newborn screening for sickle cell is required in every state in the United States. In the European Union, not all countries screen for sickle cell disease. In sub-Saharan Africa, there is very little screening.
Blood Test
For children and adults who are unsure if they have the sickle cell gene or any abnormal hemoglobin, they can find out with a simple blood test, provided by a healthcare provider. A hemoglobin electrophoresis test can determine if an abnormal hemoglobin gene is present, however, if a rare abnormal hemoglobin gene is detected, additional tests may be needed to confirm genetic status.
Treatments
Treatment options for sickle cell disease vary based on available resources and the associated complications. Below are some commonly recommended preventative measures and treatments for sickle cell disease complications.
Patients are highly advised to consult with their primary care provider or hematologist to ensure which treatment option, preventative option, and clinical studies are right for them.
| Treatment Options | Treatment Name | Purpose/Description |
| Preventative | Folate/Folic Acid | This B vitamin is important for those with sickle cell disease because it targets anemia by working to make new red blood cells. |
| Antibiotics | Antibiotics destroy bacteria that enter the body and further prevent potential complications that result from infections. Babies diagnosed with SCD should begin taking penicillin as early as two months of age. While penicillin does not prevent infections alone, it prolongs the initial infectious stage that creates a fever, which gives time for the parent/caregiver to seek medical attention for proper diagnosis and treatment. | |
| Vaccination | Vaccines also prevent infections or help to reduce the possibility of severe complications due to an infection. The two important vaccines to note are for Haemophilus influenzae (Hib vaccine) and Streptococcus pneumoniae (Pneumococcal vaccine). Be sure to stay up to date with your vaccinations! | |
| Blood Transfusions | Simple Blood Transfusion: a simple blood transfusion adds healthy blood to the body to decrease the percent of sickle cells. Chronic Blood Transfusion: transfusions that are scheduled every two to four weeks. This therapy is usually for those with sickle cell disease that have a high risk of stroke or have had other life-threatening complications of sickle cell disease. Exchange transfusion: the patient’s blood is removed and replaced with healthy, normal donor red blood cells. This can be done manually by a nurse or with a machine that performs an automated red cell exchange. | |
| Pain Management | NSAIDs | NSAIDs, or non-steroidal anti-inflammatory drugs relieve pain and reduce inflammation that can also create pain in the body. They are available over the counter. Examples: Motrin/ ibuprofen, Tylenol/acetaminophen |
| Opioids | Opioids are a class of drugs used to reduce or relieve pain. They are only available by prescription. Examples: Dilaudid/hydromorphone, OxyContin/oxycodone | |
| Alternative pain management | Heat therapy, meditation, visualization, distraction, transcutaneous electrical nerve stimulation, cannabis | |
| FDA-Approved Pharmaceuticals | Hydroxyurea | Hydroxyurea, or hydrea, boosts the production of fetal hemoglobin, which prevents the formation of sickled cells. Hydroxyurea was FDA approved in 1998 and is a recommended treatment option for sickle cell disease. The FDA approved hydroxyurea for children as young as 2 years in 2017. Some centers that start hydroxyurea earlier than two years. |
| L-glutamine | L-glutamine helps to manage and prevent damage in red blood cells and works to reduce the number of sickle cell crises in adults and children older than age five. L-glutamine was FDA approved in 2017. | |
| Adakveo (crizanlizumab-tmca) | Adakveo is a long-term, IV infusion treatment used to reduce the frequency of pain vaso-occlusive crises in adults and pediatric patients ages 16 and older. Adakveo was FDA approved in 2019. | |
| Oxbryta (Voxelotor) | Oxbryta prevents red blood cells from transforming into sickled red blood cells because it holds some of the hemoglobin in their oxygenated position, which prevents sickling in those with HbSS particularly. This decreases hemolysis and anemia and increases oxygen delivery to the body. Oxbryta was recently FDA approved in 2019 for ages 12 and older. | |
| Curative | Bone Marrow/Stem Cell Transplant | This procedure is currently the only known cure for sickle cell disease. An HLA-matched donor, usually a sibling, is required. However, there are ongoing studies in adults to use a relative who is a “half” match, like a parent, for the stem cells. |
| Gene Therapy |
In developing countries, over 80% of babies born with sickle cell anemia die within the first 5years of life, with the highest incidence of stroke occurring between age 2-5years. So many people have lived a healthy long life with sickle cell anemia, with a proper understanding of the disease, avoiding trigger factors, proper diet, and hydration, proper vaccines especially against encapsulated bacteria due to auto splenectomy which occurs in childhood and with proper medication, oxygen, during sickle cell crisis. Sickle cell anemia can be prevented by proper education, awareness and premarital counselling.
Sickle Cell Date to remember
Thanks to the significant early efforts of Madame Edwidge Ebakisse Badassou from the Democratic Republic of Congo and Madame Antoinette Sassou N’Guesso from the Democratic Republic of Congo, and Madame Viviane Wade from Senegal, sickle cell disease is received greater awareness and is recognized by the World Health Organization as a public health problem that requires research, awareness, and annual recognition.
Today, we observe Sickle Cell Disease and those affected by the disease annually.
| Date/Month of Observance | Observance Name |
| June 19 | World Sickle cell Day |
| July | UK National Sickle Cell Awareness Month |
| September | U.S. National Sickle Cell Awareness Month |
Sickle cell and Mental Health
The prevalence of sickle cell anemia has continued to grow over the years, but unfortunately the psychological effect of the disease has received little to no attention in the clinical and research setting. Most scholar articles on sickle cell anemia, focuses on the physiological aspect of the disease with no education on the psychology. Sickle cell warriors are faced with various degree of psychological impact in different aspect of their lives; education, occupation, finance, family, and social organization. Over the years people living with sickle cell disease have struggled with addiction to opioids medication due to chronic pain, financial crisis due to frequent hospitalization, low self-esteem due to stigmatization and daily person struggles.
Stigmatization exists in different societies and it’s a constant concern and struggle of a cell warrior and generally with patients of chronic illness.
In Nigeria for example some cultures believed sickle cell was a form of witchcraft, curse, punishment for your sins or that of your parents. This led to isolation of sickle cell warriors which caused increase emotional instability.
Although not as prevalent because of the increasing knowledge of the disease, and general education, it’s still a cause of concern as they are still a large number of people with little to no knowledge of the disease and as human beings, we tend to avoid what we do not understand.
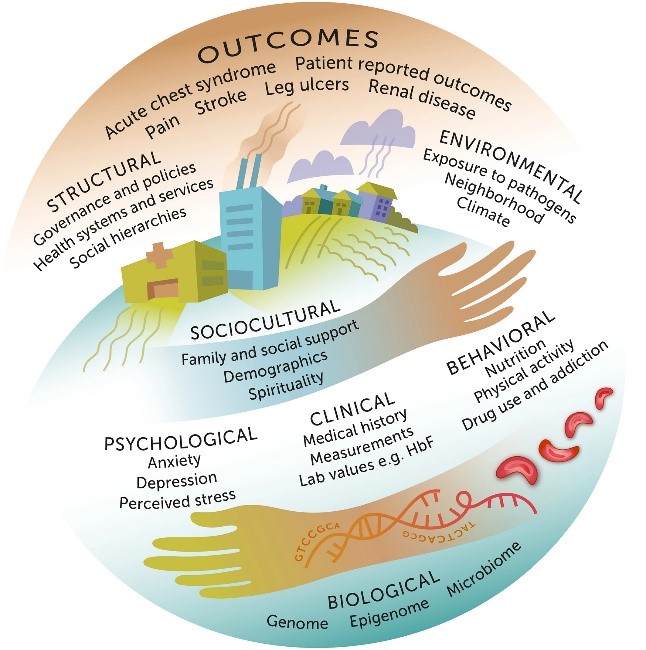
As with most chronic disease, depression and psychiatric disorders are common in sickle cell warriors. The psychological impact on cell warrior also extend to their families as frequent hospitalization, treatment and care can become a burden both emotionally and financially. Children with sickle cell anemia are at increased risk of suffering from depression and emotional disorders due to the lack of proper coping mechanisms, increase guilt and anger. Repeated hospital visits leading to absent school days, inability to experience a normal childhood and most of their activities being restricted. Children begin blaming themselves or their parents which can lead to increase sadness and eventually increase pain crises.
Guilt is a familiar feeling among people living with sickle cell anemia as they believe that the reason for their illness is them, having to question if they’re well hydrated or how much stress one can take, failing to realize sometimes your cells just sickle. In a society where stigmatization is prevalent, living with sickle cell becomes harder as many try to hide the fact that they have the disease to avoid being labelled.
Management
Psychoeducational interventions: This focuses on educating the warriors, providing an in-depth of the disease, and psychological support. Warriors with better knowledge of the disease have been shown to have a suitable coping method during painful crises and stressful situations.
Group therapy: The idea with group therapy is to create a familiar environment, where warriors can meet other warriors and feel less isolated. Warriors can also gain support and encouragement through shared experiences. Family therapy can also be beneficial in providing member of the family with a better knowledge of the disease and helping them cope through the financial stress and frequent hospitalization as this can cause a strain in the family.
Good physician-patient relationship can also provide a comfortable environment, where patients don’t feel judged, or labelled.
Other coping methods; healthy relationships, spending time with family and friends, finding new hobbies and doing the things that you love, having a support system, activities that help take your mind of the condition and always having someone to talk to is very important and can reduce the rate of depression and emotional disorders. These methods can also help with other forms of chronic pain disorder.